class: split-60 title-slide2 with-border white background-image: url("images/bg2.jpg") background-size: cover .column.shade_main[.content[ <br><br> # Statistical Methods for Omics Assisted Breeding ## <b style='color:#FFEB3B'>Introduction to ASReml-R</b> ### <br>Emi Tanaka <br>emi.tanaka@sydney.edu.au <br>School of Mathematics and Statisitcs ### 2018/11/14 <br> ### <span style='font-size:14pt; color:pink!important'>These slides may take a while to render properly. You can find the pdf <a href="https://www.dropbox.com/s/9vn57v9sls7jtwu/day3-session01-intro-to-asreml.pdf?dl=1">here</a>.</span> <br><br> ### <a rel='license' href='http://creativecommons.org/licenses/by-sa/4.0/'><img alt='Creative Commons License' style='border-width:0; width:30pt' src='images/cc.svg' /><img alt='Creative Commons License' style='border-width:0; width:20pt' src='images/by.svg' /><img alt='Creative Commons License' style='border-width:0; width:20pt' src='images/sa.svg' /></a><span style='font-size:10pt'> This work by <span xmlns:cc='http://creativecommons.org/ns#' property='cc:attributionName'>Emi Tanaka</span> is licensed under a <a rel='license' href='http://creativecommons.org/licenses/by-sa/4.0/'>Creative Commons Attribution-ShareAlike 4.0 International License</a>.</span> ]] .column[.content[ ]] <img src="images/USydLogo-white.svg" style="position:absolute; top:80%; left:40%;width:200px"> --- class: split-70 with-border .column[.content[ # Linear Mixed Model <center> <img src="images/mixedmodeleqn.png" width="40%"/> </center> <center> <img src="images/mmassumption.png" width="40%"/> </center> Estimate of fixed effects and prediction of random effects is given by: <center> <img src="images/blue.png" width="50%"/> </center> ]] .column.bg-blue[.content[ .center[ # .font_large[.font_large[<span><i class="fas fa-book faa-none animated "></i></span>]] ] .font_small[ Source: [Morota (2015) The Origin of BLUP and MME.](http://morotalab.org/literature/2015/03/07/The-Origin-of-BLUP-and-MME/)<br> **First appearance of MME and BLUP:** * Henderson (1949) Estimation of changes in herd environment. *Journal of Dairy Science (Abstract)* **32** 706 * Henderson (1950) Estimation of genetic parameters. *Ann Math Stat (Abstract)* **21** 309-310 **The formal proof that solutions in Henderson (1949, 1950) are BLUE and BLUP:** * Henderson et al. (1959) The Estimation of environmental and genetic trends from records subject to culling. *Biometrics* **15** 192–218 * Henderson (1963) Selection index and expected genetic advance. *In Statistical Genetics and Plant Breeding* 141-163. NAS-NRC 982, Washington, DC ]]] --- class: split-70 with-border .column[.content[ # A brief history of ASReml * The core algorithm of ASReml by Gilmour et al. (2002) is written in Fortran. * `asreml` R package by Butler et al. (2009) wraps the core algorithm. <img src="images/asreml.png" width="100%"> * ASReml uses REML (Patterson and Thompson, 1971) and AI algorithm and sparse matrix methods (Gilmour et al. 1995) to fit the specified (generalised) .indigo[linear mixed models]. ]] .column.bg-blue[.content[ .center[ # .font_large[.font_large[<span><i class="fas fa-book faa-none animated "></i></span>]] ] .font_small[ * **Seminal paper for REML**: Patterson & Thompson (1971) Recovery of inter-block information when block sizes are unequal. *Biometrika* **58**(3) 545-554 * **First appearance of AI algorithm**: Johnson & Thompson (1995) Restricted Maximum Likelihood Estimation of Variance Components for Univariate Animal Models Using Sparse Matrix Techniques and Average Information. *Journal of Dairy Science* **78**(2) 449-456 * **AI algorithm in ASReml**: Gilmour et al. (1995) Average Information REML: An Efficient Algorithm for Variance Parameter Estimation in Linear Mixed Models. *Biometrics* **51**(4) 1440-1450 * Gilmour et al. (2002) ASReml User Guide. Release 1.0. VSN International Ltd, Hemel Hempstead, HPI 1 ES, UK. * Butler et al. (2009) Mixed models for S language environments: ASReml-R reference manual. Version 3. VSN International Ltd, Hemel Hempstead, HPI 1 ES, UK. Also read [Morota (2015) Variance Component Estimation.](http://morotalab.org/literature/2015/01/19/Variance-Component-Estimation/) ]]] --- class: split-60 with-border .column[.content.vmiddle[ # <span><i class="fas fa-exclamation-triangle faa-flash animated " style=" color:red;"></i></span> # asreml-R version 3 ## The syntax used are for version 3. ## Version 4 have some different syntax which we do not cover today but details can be found [here](https://asreml.kb.vsni.co.uk/wp-content/uploads/sites/3/2018/07/Navigating-from-ASReml-R-3-to-4.pdf.pdf). ]] .column.bg-blue.font_sm90[.content.vmiddle[ # References: * Butler et al. (2017) Navigating from ASReml-R Version 3 to 4. [Technical Report.](https://asreml.kb.vsni.co.uk/wp-content/uploads/sites/3/2018/07/Navigating-from-ASReml-R-3-to-4.pdf.pdf). * Butler et al. (2018) ASReml-R Reference Manual Version 4. [Technical Report.](https://asreml.kb.vsni.co.uk/wp-content/uploads/sites/3/2018/02/ASReml-R-Reference-Manual-4.pdf) ]] --- class: split-70 with-border .column[.content[ # Illustrative Example<sup>*</sup>: Multi-environmental Field Trial <img src="day3-session01-intro-to-asreml_files/figure-html/eg1-1.png" style="display: block; margin: auto;" /><img src="day3-session01-intro-to-asreml_files/figure-html/eg1-2.png" style="display: block; margin: auto;" /> .bottom_abs.width100.font_small[ <sup>*</sup>That is, unrealistic example only used to illustrate the basic idea. ] ]] .column.bg-blue[.content[ ### <u>Data</u> * 3 Genotypes *j* * 3 Sites *i* * 2 Replicates *k* * Response: Yield *y<sub>ijk</sub>* ### <u>Proposed Model</u><sup>*</sup> <img src="images/model1.png" width="97%"/> We show how to fit this model with different variance structures. .bottom_abs.width100.font_small[ <sup>*</sup>This is not to say this is the best model! ]]] --- class: split-60 with-border .column[.content[ # Model Fitting in `asreml` ```r asreml(fixed=response ~ fixed formula, random= ~ random formula, rcov= ~ rcov formula, data=data.frame) ``` <img src="images/model1v2.png" width="100%"/> ## Model 1: Simple Model <center> <img src="images/assum1.png" width="90%"/> </center> ]] .column.bg-blue[.content[ ```r head(data1) ``` ``` # A tibble: 6 x 5 Row Col Site Geno Yield <fct> <fct> <fct> <fct> <dbl> 1 1 1 A G1 5.14 2 2 1 A G3 7.15 3 1 2 A G2 3.80 4 2 2 A G2 6.41 5 1 3 A G1 4.30 6 2 3 A G3 6.91 ``` To fit Model 1 in `asreml`: ```r fit1 <- asreml(Yield ~ Site, random= ~ Site:Geno, data=data1) ``` ]] --- class: split-90 with-border .column[ .split-two[ .row[.content[ # `asreml` output ## Variance Parameter Estimate ```r summary(fit1)$varcomp # or use lucid::vc(fit1) ``` ``` gamma component std.error z.ratio constraint Site:Geno!Site.var 1.093095 1.0663211 0.9262377 1.151239 Positive R!variance 1.000000 0.9755063 0.4598581 2.121320 Positive ``` ]] .row[.content[ .split-two[ .column[.content[ ## E-BLUE ```r coef(fit1)$fixed ``` ``` effect Site_A 0.0000000 Site_B -1.0396488 Site_C 0.1556465 (Intercept) 5.6176863 ``` ]] .column[.content[ ## E-BLUP ```r coef(fit1)$random ``` ``` effect Site_A:Geno_G1 -0.6172425 Site_A:Geno_G2 -0.3519268 Site_A:Geno_G3 0.9691694 Site_B:Geno_G1 0.2607835 Site_B:Geno_G2 -0.9825827 Site_B:Geno_G3 0.7217992 Site_C:Geno_G1 -0.6441000 Site_C:Geno_G2 -0.2980348 Site_C:Geno_G3 0.9421349 ``` ]] ]] ] ]] .column.bg-blue[.content[ ]] --- class: split-70 with-border .column[.content[ ## Model 1 continued `fit1` is equivalent to ```r fit1a <- asreml(Yield ~ Site, data=data1 * random= ~ idv(Site):id(Geno)) fit1b <- asreml(Yield ~ Site, data=data1, * random= ~ id(Site):idv(Geno)) ``` where the .indigo[variance structures of random effects] are made explicit. Additionally we can define the .indigo[residual error variance structure] explicit: ```r fit1c <- asreml(Yield ~ Site, data=data1, random= ~ Site:Geno, * rcov= ~ idv(units)) ``` where `units` is a special reserve word in `asreml` equivalent to `factor(1:nrow(data1))`. ]] .column.bg-blue[.content.font_sm80[ # <span><i class="fas fa-info-circle faa-none animated " style=" color:white;"></i></span> .white[Defaults] * By default, `asreml` has an `idv` structure if the variance structure is not defined. E.g. `random=~Site` means <center> <img src="images/eg1.png" width="80%"> </center> * If `rcov` is not defined, the default is `idv(units)`. * For random interaction effects (say `Site:Geno`), the default is `idv(Site):id(Geno)` (or equivalently `id(Site):idv(Geno)`) <center> <img src="images/eg2.png" width="100%"> </center> ]] --- class: split-50 with-border .column[.content[ # Variance vs. Correlation Structure * `id()` is a correlation structure `\(\boldsymbol{I}_n\)` * `idv()` is a variance structure with common (homogeneous) variance `\(\sigma^2\boldsymbol{I}_n\)` * `idh()` (which is the same as `diag()`) is a variance structure with a separate (heterogeneous) variance for each level of factor `\(\text{diag}(\sigma^2_1, ..., \sigma^2_n)\)`. Appending the name of the correlation function with: * `v` results in adding a homogeneous variance or * `h` results in heterogeneous variance. ]] .column.bg-blue[.content.pad10px[ ## The full list of pre-defined correlation structures ## <u>Time Series Type</u> | <u>Metric Based</u> | <u>General</u> ------------- | ------------- | ------------- `ar1` | `exp` | `cor` `ar2` | `gau` | `corb` `ar3` | `iexp` | `corg` `sar` | `igau` | `id` `sar2` | `ieuc` | `us` `ma1` | `sph` | `chol` `ma2` | `cir` | `cholc` `arma` | `aexp` | `ante` | `agau` | `fa` | `mtrn` | `rr` ]] --- class: split-70 with-border .column[.content[ # Separable Structure * Given a random term `A:B` where `A` and `B` are factors then the generic form is `f(A):g(B)` where * `f(A)` generates a variance matrix **G**<sub>A</sub> say, and * `g(B)` generates a variance matrix **G**<sub>B</sub> say. Then the corresponding random effects, <img src="images/Uab.png" style="height:0.8em"/>, has properties: * <img src="images/varUab.png" style="height:0.85em"/>; and * <img src="images/covgagb.png" style="height:0.9em"/>. * Note assumption of separability * differs to assuming a completely unstructured variance <img src="images/vargab.png" style="height:0.8em"/>; and * greatly reduces computational load. ]] .column.bg-blue[.content[ # `\(\otimes\)` Kronecker Product Given general matrices: <img src="images/Amat.png" width="80%"/> and **B**. Then <img src="images/eg3.png" width="90%"/> Note: *g<sub>A<sub>i,j</sub></sub>* means the *(i,j)*-th entry of matrix **G**<sub>A</sub>. Similar definition for *g<sub>B<sub>k,l</sub></sub>*. ]] --- class: split-50 with-border .column[.content[ # Model 2: MET Unstructured Model <img src="images/assum2.png" width="100%"/> * Same genotype across sites * General idea: *borrow strength* across sites to get more accurate `Site:Geno` prediction. ]] .column.bg-blue[.content[ ```r fit2 <- asreml(Yield ~ Site, random=~ us(Site):id(Geno), data=data1) ``` <center> <img src="images/assum2v2.png" width="65%"/> </center> .font_sm80.pad1[Note: below value is *rubbish*. More on this later.] ```r summary(fit2, nice=T)$nice ``` ``` $`Site:Geno` A B C A 0.1500000 0.2209702 0.663163760 B 0.2209702 0.4615557 0.155530075 C 0.6631638 0.1555301 0.009486833 ``` ]] --- class: split-50 with-border .column[.content[ ```r summary(fit2, all=T) %>% str() ``` ``` List of 9 $ call : language asreml(fixed = Yield ~ Site, random = ~us(Site):id(Geno), data = data1) $ loglik : num -13.4 $ nedf : int 15 $ sigma : num 1.18 $ varcomp :'data.frame': 7 obs. of 5 variables: ..$ gamma : num [1:7] 0.15 0.221 0.462 0.663 0.156 ... ..$ component : num [1:7] 0.209 0.308 0.644 0.925 0.217 ... ..$ std.error : num [1:7] 0.118 0.593 0.733 0.846 0.592 ... ..$ z.ratio : num [1:7] 1.774 0.52 0.878 1.093 0.366 ... ..$ constraint: Factor w/ 3 levels "?","Positive",..: 3 2 2 2 2 1 2 .. ..- attr(*, "names")= chr [1:7] "Site:Geno!Site.A:A" "Site:Geno!Site.B:A" "Site:Geno!Site.B:B" "Site:Geno!Site.C:A" ... $ distribution: chr "gaussian" $ link : chr "identity" $ coef.fixed : num [1:4, 1:3] 0 -1.04 0.156 5.618 NA ... ..- attr(*, "dimnames")=List of 2 .. ..$ : chr [1:4] "Site_A" "Site_B" "Site_C" "(Intercept)" .. ..$ : chr [1:3] "solution" "std error" "z ratio" $ coef.random : num [1:9, 1:3] -0.238 -0.301 0.539 -0.121 -0.385 ... ..- attr(*, "dimnames")=List of 2 .. ..$ : chr [1:9] "Site_A:Geno_G1" "Site_A:Geno_G2" "Site_A:Geno_G3" "Site_B:Geno_G1" ... .. ..$ : chr [1:3] "solution" "std error" "z ratio" - attr(*, "class")= chr "summary.asreml" ``` ]] .column.bg-blue[.content[ ```r summary(fit2, all=T) ``` ``` $call asreml(fixed = Yield ~ Site, random = ~us(Site):id(Geno), data = data1) $loglik [1] -13.40281 $nedf [1] 15 $sigma [1] 1.180882 $varcomp gamma component std.error z.ratio constraint Site:Geno!Site.A:A 0.150000000 0.20917239 0.1178934 1.77424976 Singular Site:Geno!Site.B:A 0.220970186 0.30813908 0.5925541 0.52001846 Positive Site:Geno!Site.B:B 0.461555711 0.64363140 0.7334553 0.87753330 Positive Site:Geno!Site.C:A 0.663163760 0.92477032 0.8461568 1.09290650 Positive Site:Geno!Site.C:B 0.155530075 0.21688398 0.5919423 0.36639377 Positive Site:Geno!Site.C:C 0.009486833 0.01322922 1.1282792 0.01172513 ? R!variance 1.000000000 1.39448259 0.7859562 1.77424976 Positive $distribution [1] "gaussian" $link [1] "identity" $coef.fixed solution std error z ratio Site_A 0.0000000 NA NA Site_B -1.0396488 0.7150596 -1.4539330 Site_C 0.1556465 0.7150596 0.2176692 (Intercept) 5.6176863 0.5496707 10.2200938 $coef.random solution std error z ratio Site_A:Geno_G1 -0.2377406 0.4021136 -0.5912274 Site_A:Geno_G2 -0.3010980 0.4021136 -0.7487883 Site_A:Geno_G3 0.5388385 0.4021136 1.3400157 Site_B:Geno_G1 -0.1214088 0.4021136 -0.3019265 Site_B:Geno_G2 -0.3846551 0.4021136 -0.9565832 Site_B:Geno_G3 0.5060639 0.4021136 1.2585097 Site_C:Geno_G1 -0.2412990 0.4021136 -0.6000766 Site_C:Geno_G2 -0.2939577 0.4021136 -0.7310314 Site_C:Geno_G3 0.5352567 0.4021136 1.3311081 attr(,"class") [1] "summary.asreml" ``` ]] --- class: split-50 with-border .column[.content[ # Model 3: G-BLUP Model * Genotypes are often related with its relatedness inferred from .indigo[pedigree] or .indigo[genetic markers]. * This *known* structure (denoted `\(\textbf{G}_g\)` here) is fitted in the variance structure as follows: <img src="images/assum4.png" width="80%"/><br> Say, <br> <img src="images/assum4add.png" width="100%"/> ]] .column.bg-blue[.content[ ```r fit3 <- asreml(Yield ~ Site, data=data1, random=~ us(Site):giv(Geno), ginverse=list(Geno=Ginv)) ``` where `Ginv` is: ```r Ginv ``` ``` Row Column Value 1 1 1 1.5 2 2 1 0.5 3 2 2 1.5 4 3 1 -1.0 5 3 2 -1.0 6 3 3 2.0 ``` ```r attr(Ginv, "rowNames") ``` ``` [1] "G1" "G2" "G3" ``` ]] --- class: split-50 with-border .column[.content[ # Model 4: Multi-Trait MET Model * It is perhaps more realistic to fit a model where the residual error variance differ at different trials. <center> <img src="images/assum5.png" width="50%"/> </center> * `at(f,l)` conditions on level `l` of factor `f`. If `l` is not specified, conditions on each level of `f`. ]] .column.bg-blue[.content[ ```r fit4 <- asreml(Yield ~ Site, data=data1, random=~ us(Site):giv(Geno), ginverse=list(Geno=Ginv), * rcov=~at(Site):units) ``` Rules for residual error term: 1. The number of effects in `rcov` must be equal to the number of obeservational units included in the analysis. 2. Where a compound model term is specified in `rcov`, each combination of levels of the simple model terms comprising this term must uniquely identify one unit of the data. 3. The data must be ordered to match the `rcov` specified. ]] --- class: split-60 with-border .column[.content[ # Analysis on Subset of Data ```r fit4A1 <- asreml(Yield ~ Site, random=~Site:Geno, * data=data1, subset=Site=="A") ``` ```r *fit4A2 <- asreml(Yield ~ 1, random=~ Geno, data=data1, subset=Site=="A") ``` ```r dataA <- subset(data1, Site=="A") fit4A3 <- asreml(Yield ~ Site, random=~Site:Geno, * data=dataA) ``` ### Residuals ```r resid(fit4A1) ``` ]] .column.bg-blue[.content[ A quick check to see if the (deviance) residual are the same: ```r all.equal(resid(fit4A1), resid(fit4A2)) ``` ``` [1] TRUE ``` ```r all.equal(resid(fit4A2), resid(fit4A3)) ``` ``` [1] TRUE ``` ```r all.equal(resid(fit4A1), resid(fit4A3)) ``` ``` [1] TRUE ``` ]] --- class: split-60 with-border .column[.content[ ```r fit4A <- asreml(Yield ~ 1, random=~ Geno, data=data1, subset=Site=="A") fit4B <- asreml(Yield ~ 1, random=~ Geno, data=data1, subset=Site=="B") fit4C <- asreml(Yield ~ 1, random=~ Geno, data=data1, subset=Site=="C") fit4 <- asreml(Yield ~ Site, data=data1, random=~ diag(Site):Geno, # idh(Site):Geno rcov=~ at(Site):units) ``` ```r lucid::vc(fit4) ``` ``` effect component std.error z.ratio constr Site:Geno!Site.A.var 0.9034 1.618 0.56 P Site:Geno!Site.B.var 1.349 1.669 0.81 P Site:Geno!Site.C.var 0.9461 1.54 0.61 P Site_A!variance 1.261 1.029 1.2 P Site_B!variance 0.6029 0.4922 1.2 P Site_C!variance 1.063 0.868 1.2 P ``` ]] .column.bg-blue[.content[ ### Equivalence of Single site and MET analysis ```r lucid::vc(fit4A) ``` ``` effect component std.error z.ratio constr Geno!Geno.var 0.9034 1.618 0.56 P R!variance 1.261 1.029 1.2 P ``` ```r lucid::vc(fit4B) ``` ``` effect component std.error z.ratio constr Geno!Geno.var 1.349 1.669 0.81 P R!variance 0.6029 0.4922 1.2 P ``` ```r lucid::vc(fit4C) ``` ``` effect component std.error z.ratio constr Geno!Geno.var 0.9461 1.54 0.61 P R!variance 1.063 0.868 1.2 P ``` ]] --- class: split-50 with-border .column[.content[ # Model 5: Spatial Model * It is perhaps more realistic to fit a model such that plots that are closer together geographically are more correlated than plots further away from each other. * We capture this by considering an autoregressive of order 1 structure for row and column direction in the field. ## Model 6: MET Factor Analytic Model * In practice, we replace the unstructured variance models with factor analytic models. More explanations on these models later today. ]] .column.bg-blue[.content[ ```r fit5 <- asreml(Yield ~ Site, data=data1, random=~ us(Site):giv(Geno), ginverse=list(Geno=Ginv), * rcov=~ at(Site):ar1v(Row):ar1(Col)) ``` <center> <img src="images/assum5v2.png" width="70%"/> </center> ```r fit6 <- asreml(Yield ~ Site, data=data1, * random=~ fa(Site, 3):giv(Geno), ginverse=list(Geno=Ginv), rcov=~ at(Site):ar1v(Row):ar1(Col)) ``` ]] --- class: split-50 with-border .column[.content[ ## Model 7: MPI Model * We can add multiple (crossed) random effects and multiple `giv`s. Here the model is fitting .indigo[additive marker] effects, .indigo[residual additive effects] (via pedigree), and .indigo[non-additive effects] (capturing interaction effects). ## Model 8: Multivariate/Multi-trait Model * Suppose you record multiple traits for each genotype and you wish to jointly model these traits. * `asreml` has a reserve word `trait` which is a factor with levels corresponding to each trait. * Notice this model is similar to the Multi-trait MET Model. ]] .column.bg-blue[.content[ ```r fit7 <- asreml(Yield ~ Site, data=data1, random=~ fa(Site, 3):giv(Geno) + * fa(Site, 2):giv(GenoDup) + * fa(Site, 2):ide(Geno), ginverse=list(Geno=Kinv, GenoDup=Ainv), rcov=~ at(Site):ar1v(Row):ar1(Col)) ``` ```r fit8 <- asreml(c(y1,y2) ~ trait, data=data1, random=~ us(trait):giv(Geno), ginverse=list(Geno=Ginv), rcov=~ units:us(trait)) ``` ]] --- class: split-50 with-border .column[.content[ # Example: Split-Plot Field Trial for Oats 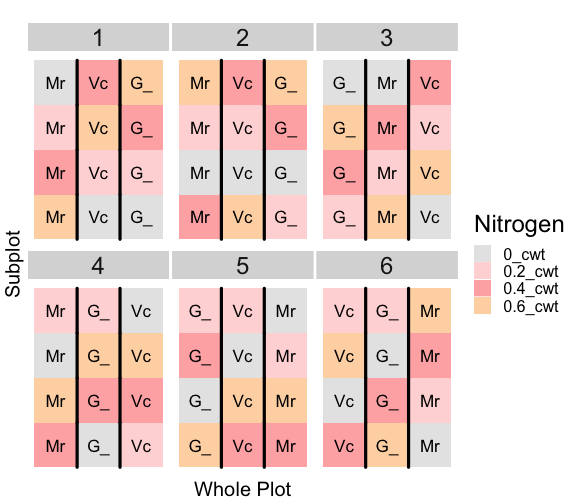<!-- --> .bottom_abs.width100.font_small[ Yates, F. (1935) Complex experiments. *Journal of the Royal Statistical Society Suppl.* **2** 181–247. ] ]] .column.bg-blue[.content[ ```r data(oats, package="asreml") # load data oats <- oats %>% mutate(Row=as.numeric(Wplots), Col=as.numeric(Subplots)) desplot(Nitrogen ~ Row*Col|Blocks, data=oats, text=Variety, cex=1.5, out1=Wplots, gg=TRUE) + labs(title="", x="Whole Plot", y="Subplot") + theme(axis.title=element_text(size=20), legend.text=element_text(size=16), legend.title=element_text(size=24), strip.text=element_text(size=24)) ``` * 3 Varieties applied to Whole Plot * Nitrogen applied to Subplot within Whole Plot * **Question**: do Nitrogen and/or Variety have an affect on yield? ]] --- class: split-90 with-border .column[.content[ # Inference for fixed effects: Wald Test ```r fit <- asreml(yield ~ Variety*Nitrogen, random= ~ Blocks/Wplots, data=oats) wald(fit) # same as anova(fit) ``` ``` Wald tests for fixed effects Response: yield Terms added sequentially; adjusted for those above Df Sum of Sq Wald statistic Pr(Chisq) (Intercept) 1 43410 245.141 <2e-16 *** Variety 2 526 2.971 0.2264 Nitrogen 3 20020 113.057 <2e-16 *** Variety:Nitrogen 6 322 1.817 0.9357 residual (MS) 177 --- Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1 ``` .font_small.width100.font_small[ Wald (1943) Tests of Statistical Hypotheses Concerning Several Parameters When the Number of Observations is Large. *Transactions of the American Mathematical Society* **54**(3) 426-482 ] ]] .column.bg-blue[.content[ ]] <img src="images/wald.png" width="40%" style="position:absolute;top:25%;left:58%; background-color:rgba(0,0,0,0.3);padding:10px"/> --- class: split-75 with-border .column[.content[ # Kenward-Roger approximation to Wald Test * Variance parameters are estimated thus the asymptotic results in the previous slide do not hold well. * Kenward and Roger (1997) presented a .indigo[scaled Wald statistic] with an .indigo[F approximation] to its sampling distribution and an improved estimator for the precision of fixed effects (i.e. an adjusted variance matrix). * The .indigo[numerator degrees of freedom] for each term is determined as the number of non-singular equations involved in the term. * The calculation of the .indigo[denominator degrees of freedom] is in general not trivial and is computationally expensive. * `asreml` uses a partial implementation of Kenward-Roger approach. More specifically, it uses an unadjusted variance matrix with variance parameters fixed at the REML estimates of the maximal model. .bottom_abs.width100.font_small[ Kenward, M.G. and Roger, J.H. (1997). Small Sample Inference for Fixed Effects from Restricted Maximum Likelihood. *Biometrics* **53** 983-997. ] ]] .column.bg-blue[.content[ To compute the denominator degrees of freedom in `asreml`: * `denDF="default"` * `denDF="numeric"` * `denDF="algebraic"` Default is `denDF="none"`. If denominator df is unavailable, you may use residual df (`fit$nedf`). Note this will be anti-conservative. ]] --- class: split-70 with-border .column[.content[ # Conditional Tests * By default, `asreml` show the `incremental` tests where the sum of the squares are adjusted for the terms listed above the rows. * You can switch to `conditional` tests by using the argument `ssType="conditional"`. then Nelder (1977, 1994) argues that meaningful and interesting test for terms can only be conducted for those that respect marginality relations. * For example, consider a factorial model ```r y ~ 1 + A + B + A:B ``` then `A` cannot be adjusted for `A:B`. ]] .column.bg-blue[.content[ ]] --- class: split-80 with-border .column[.content[ # Conditional Tests in `asreml` ```r out_wald <- wald(fit, ssType="conditional", denDF="numeric") ``` ```r out_wald$Wald %>% lucid::lucid() ``` ``` Df denDF F.inc F.con Margin Pr (Intercept) 1 5 245 245 0.0000193 Variety 2 10 1.48 1.48 A 0.272 Nitrogen 3 45 37.7 37.7 A 0 Variety:Nitrogen 6 45 0.303 0.303 B 0.932 ``` * `Variety:Nitrogen` is adjusted for other terms; * `Variety` is adjusted for `Nitrogen` and `(Intercept)`; and * `Nitrogen` is adjusted for `Variety` and `(Intercept)`. ]] .column.bg-blue[.content.vmiddle[ ]] --- class: split-70 with-border .column[.content[ # Reisdual likelihood ratio test ```r data("burgueno.rowcol", package="agridat") fit1 <- asreml(yield ~ gen, random=~rep, trace=F, data=burgueno.rowcol) fit2 <- asreml(yield ~ gen, trace=F, data=burgueno.rowcol) lrt(fit2, fit1) ``` ``` Likelihood ratio test(s) assuming nested random models. Chisq probability adjusted using Stram & Lee, 1994. Df LR statistic Pr(Chisq) fit1/fit2 1 21.141 2.133e-06 *** --- Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1 ``` ```r -2 * (fit2$loglik - fit1$loglik) ``` ``` [1] 21.1414 ``` ]] .column.bg-blue[.content[ * Residual likelihood ratio test is only valid if the model has the same fixed effects and the random effects are tested. * The left is testing hypothesis `\(H_0: \sigma^2_r=0\)` vs. `\(H_1: \sigma^2_r > 0\)` where variance compnent of `rep`, `\(\sigma^2_r\)`. ]] --- class: split-80 with-border .column[.content[ # Prediction ```r fit <- asreml(Yield ~ Site + Geno, data=data1, random=~ Site:Geno + at(Site, "A"):Row) *predict(fit, classify="Geno")$predictions$pval ``` ``` Notes: - The predictions are obtained by averaging across the hypertable calculated from model terms constructed solely from factors in the averaging and classify sets. - Use "average" to move ignored factors into the averaging set. - The SIMPLE averaging set: Site - The ignored set: Row Geno predicted.value standard.error est.status 1 G1 5.049309 0.4423711 Estimable 2 G2 4.529920 0.4253100 Estimable 3 G3 6.389828 0.4423711 Estimable ``` ]] .column.bg-blue.font_sm80[.content[ 1. Includes the variables in the .indigo[classify set] (`classify`). 2. Specify the .indigo[average set] (`average`) which by default is all fixed effects not in the classify set. 3. Determine terms to use (`use`, `except`, `only`) in prediction. 4. Choose weights (`average`). ]] --- class: split-60 with-border .column[.content[ # Understanding prediction calculation <iframe src="html_extras/table1.html" width="100%" height="210!important" frameBorder="0"> </iframe> * classify set: `Geno` * averaging set: `Site` using uniform weights * ignored set: `Row` ``` Geno predicted.value standard.error est.status 1 G1 5.049309 0.4423711 Estimable 2 G2 4.529920 0.4253100 Estimable 3 G3 6.389828 0.4423711 Estimable ``` ]] .column.bg-blue[.content[ ```r 0.000 + 5.344 + (0.002 + 0.138 - 0.141)/3 + (0.000 - 1.040 + 0.156)/3 ``` ``` [1] 5.049 ``` ```r -0.519 + 5.344 + (0.059 - 0.135 + 0.076)/3 + (0.000 - 1.040 + 0.156)/3 ``` ``` [1] 4.530333 ``` ```r 1.341 + 5.344 + (-0.062 - 0.003 + 0.065)/3 + (0.000 - 1.040 + 0.156)/3 ``` ``` [1] 6.390333 ``` .bottom_abs.width100.font_small[ Of course precision is lost here beyond 2 decimal places due to rounding of 3 decimal places. ] ]] --- class: split-60 with-border .column[.content[ # Understanding prediction calculation <iframe src="html_extras/table1.html" width="100%" height="210!important" frameBorder="0"> </iframe> ```r *predict(fit, classify="Geno:Site")$predictions$pval ``` ``` Site Geno predicted.value standard.error est.status 1 A G1 5.346355 0.8971775 Estimable 2 A G2 4.883918 0.8803011 Estimable 3 A G3 6.622785 0.8971775 Estimable 4 B G1 4.442765 0.4949729 Estimable 5 B G2 3.649645 0.4855925 Estimable 6 B G3 5.641703 0.4949729 Estimable 7 C G1 5.358807 0.4949729 Estimable 8 C G2 5.056196 0.4855925 Estimable 9 C G3 6.904995 0.4949729 Estimable ``` ]] .column.bg-blue[.content[ ```r 0.002 + 5.344 + 0.000 + 0.000 ``` ``` [1] 5.346 ``` ```r 0.059 + 5.344 - 0.519 + 0.000 ``` ``` [1] 4.884 ``` ```r -0.062 + 5.344 + 1.341 + 0.000 ``` ``` [1] 6.623 ``` ```r 0.138 + 5.344 + 0.000 - 1.040 ``` ``` [1] 4.442 ``` ]] --- class: split-70 with-border .column[.content[ # More information on `predict` ```r ?predict.asreml ``` And references: * Lane and Nelder (1982) Analysis of covariance and standardization as instances of prediction. *Biometrics* **38**(3) 613-621 * Gilmour et al. (2004) An efficient computing strategy for prediction in mixed linear models. *Computational Statistics and Data Analysis* **44**(4) 571-586 * Welham et al. (2004) Prediction in linear mixed models. *Australian & New Zealand Journal of Statistics* **46**(3) 325-347 ]] .column.bg-blue[.content[ ]] --- class: split-70 with-border .column[.content[ # Missing Values in `asreml` * Missing values must be encoded as `NA`. * By default, missing values in the explanatory variables cause an error: `na.method.X="fail"`. * If `na.method.X="omit"` then corresponding records with missing explanatory variables are dicarded. * By default, missing values in the response are included and estimated from the model fit: `na.method.Y="include"`. In this case a factor `mv` is created with one level for each missing response. * If `na.method.Y="omit"` then missing values in the response are discarded. * For `na.method.X="include"`, missing values in explanatory variables are replaced with zeros. *Thus it is essential that you can scale and center covariates with missing values.* ]] .column.bg-blue[.content[ Missing value treatment argument: * `na.method.X` * `na.method.Y` each with three options: * `"omit"` * `"include"` * `"fail"` ]] --- class: split-70 with-border .column[.content[ .split-40[ .row[.content[ # Construction of numerator relations matrix (**A**) * `asreml.Ainverse` uses Meuwissen and Luo (1992) to compute the inverse of the numerator relationship matrix (**A**<sup>-1</sup>) directly from the pedigree that can be supplied in `ginverse` in conjuction with `giv` or `ped`. ]] .row[ .split-40[ .column[.content[ ## Why **A**<sup>-1</sup>? * **A**<sup>-1</sup> is usually sparse (ie contains many zeros) compared to **A**; and * the algorithm only requires **A**<sup>-1</sup>. ]] .column[.content[ <iframe src="netout.html" width="100%" height="400" frameBorder="0"></iframe> ]] ]] ] ]] .column.bg-blue[.content[ ```r ped ``` ``` me mum dad fgen 1 I1 0 0 0 2 I2 0 0 0 3 I3 0 0 0 4 I4 0 0 0 5 I5 I1 I2 0 6 I6 I1 I2 0 7 I7 I1 I3 0 8 I8 I2 I3 0 9 I9 I3 I4 0 10 I10 I5 I6 0 11 I11 I7 I8 0 12 I13 I9 I9 2 ``` .bottom_abs.width100.font_small[ * Meuwissen and Luo (1992) Computing inbreeding coefficients in large populations. *Genetics Selection Evolution* **24**(305)<br> * Example from Verbyla (2011) Pedigree Technical Notes. ] ]] --- class: split-40 with-border .column[.content[ # Sparse Format of **A**<sup>-1</sup> ```r asreml.Ainverse(ped)$ginv ``` <iframe src="html_extras/table2.html" width="100%" height="400!important" frameBorder="0"> </iframe> ]] .column.bg-white[.content[ ```r asreml.Ainverse(ped)$ginv %>% # for the format for asreml asreml.sparse2mat() # to get A inverse matrix ``` <iframe src="html_extras/table3.html" width="100%" height="500!important" frameBorder="0"> </iframe> ]] --- class: split-30 with-border .column[.content[ # The **A** Matrix <iframe src="netout.html" width="100%" height="700!important" frameBorder="0"> </iframe> ]] .column.bg-white[.content[ ```r asreml.Ainverse(ped)$ginv %>% # for the format for asreml asreml.sparse2mat() %>% # to get A inverse matrix solve() # to get the A matrix ``` <iframe src="html_extras/table4.html" width="100%" height="500!important" frameBorder="0"> </iframe> ]] --- class: split-50 with-border .column[.content[ # Convergence .pad10px[ ``` LogLikelihood not converged ``` ] By default maximum iteration `maxiter` is 13 for the AI-REML algorithm. Convergence is determined by REML. You can run a few more iterations by: ```r asreml_object <- update(asreml_object) ``` or supplying the argument `maxiter` with a higher number, say 20 ```r asreml(Yield ~ Site, random=~Geno, maxiter=20) ``` ]] .column.bg-blue[.content[ Hint: ```r while(!asreml_object$converge) asreml_object <- update(asreml_object) ``` Note: this can get stuck in a loop if it never converges! **So what if the model does not seem to converge at all?** Consider decreasing the step size from default of 0.1: ```r asreml(Yield ~ Site, random=~Geno, stepsize=0.01) ``` or change to "better" initial values... ]] --- class: split-90 with-border .column[.split-30[ .row[.content[ # Initial Value * `asreml` uses .indigo[Quasi Newton-Raphson Algorithm] to solve for REML estimates. * Starting values can impact the convergence as shown in an example below. ]] .row[ .split-two[ .column[ Initial value = 1 .img-fill[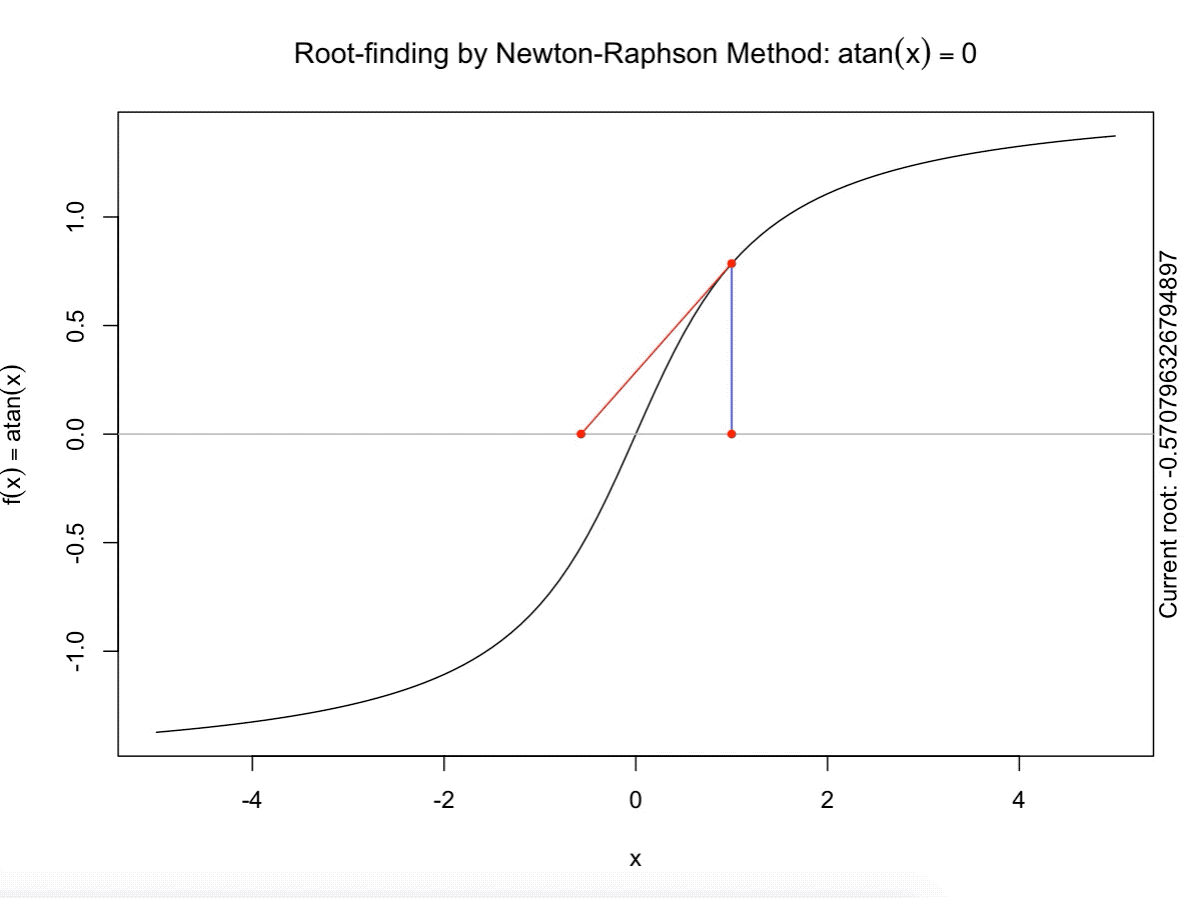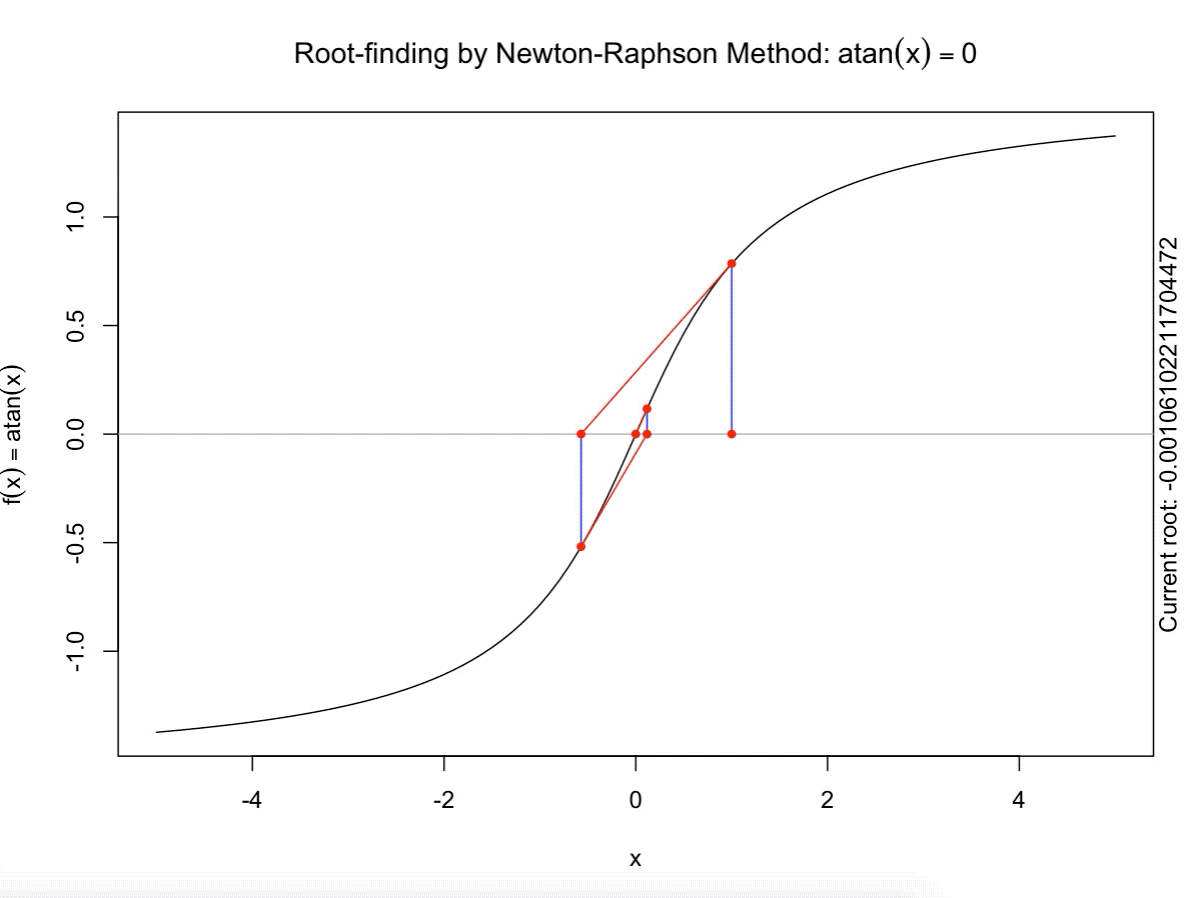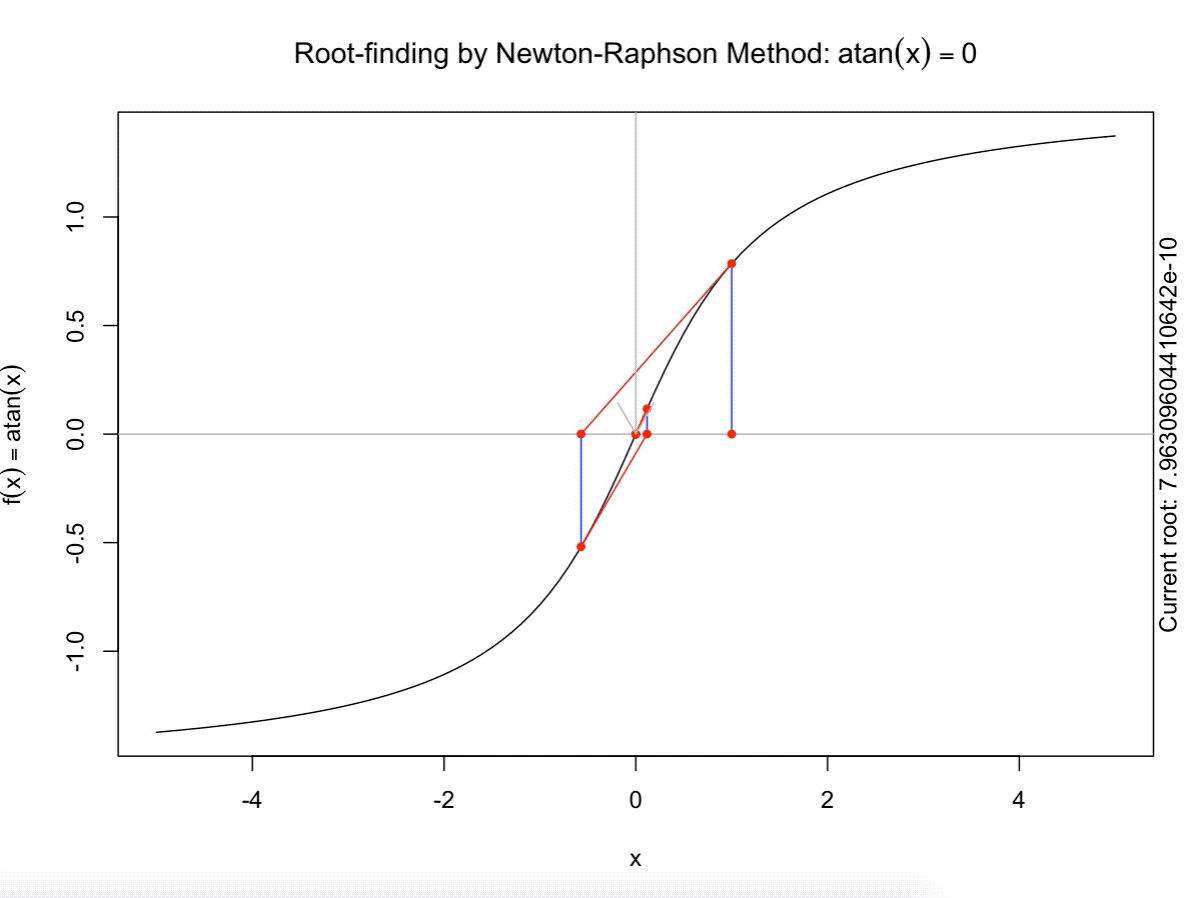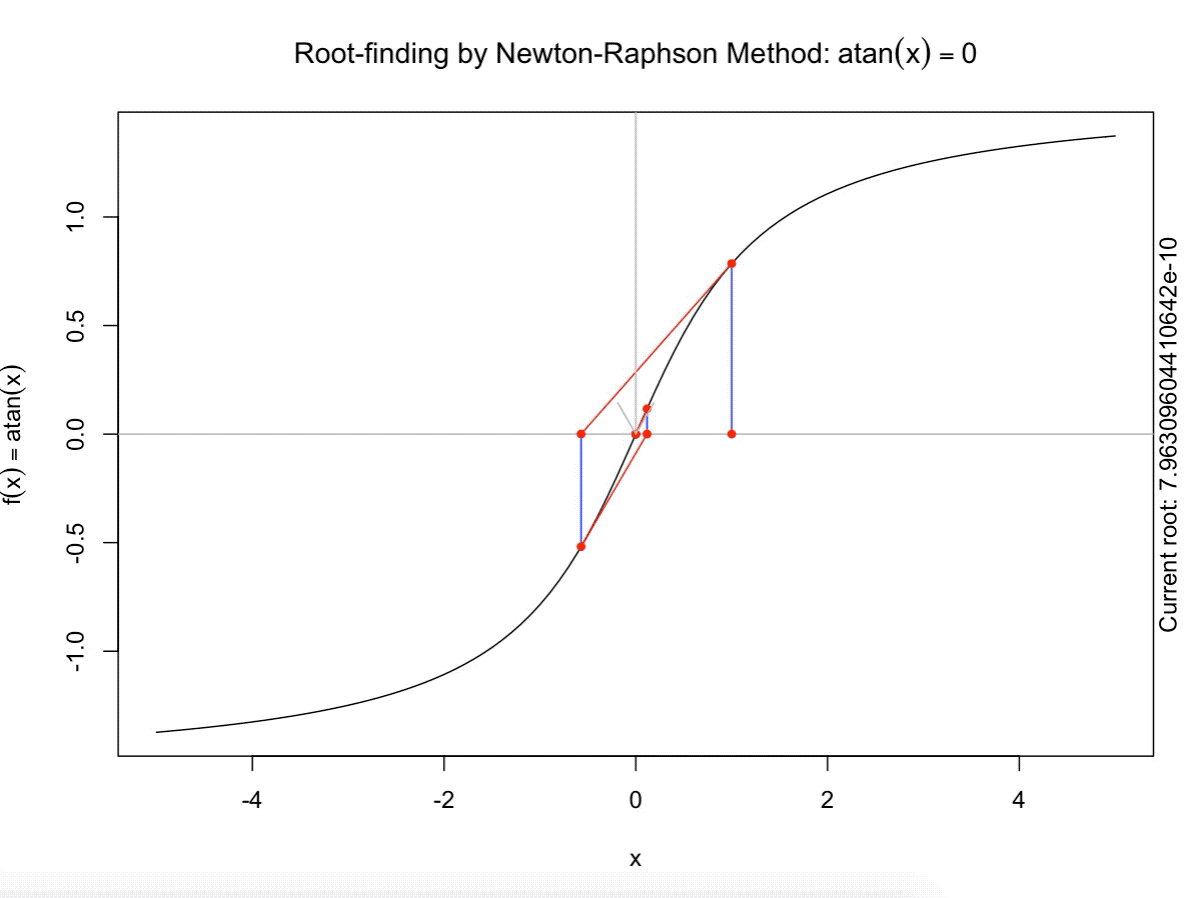] Converges to the solution *x=0*. ] .column[ Initial value = 1.5 .img-fill[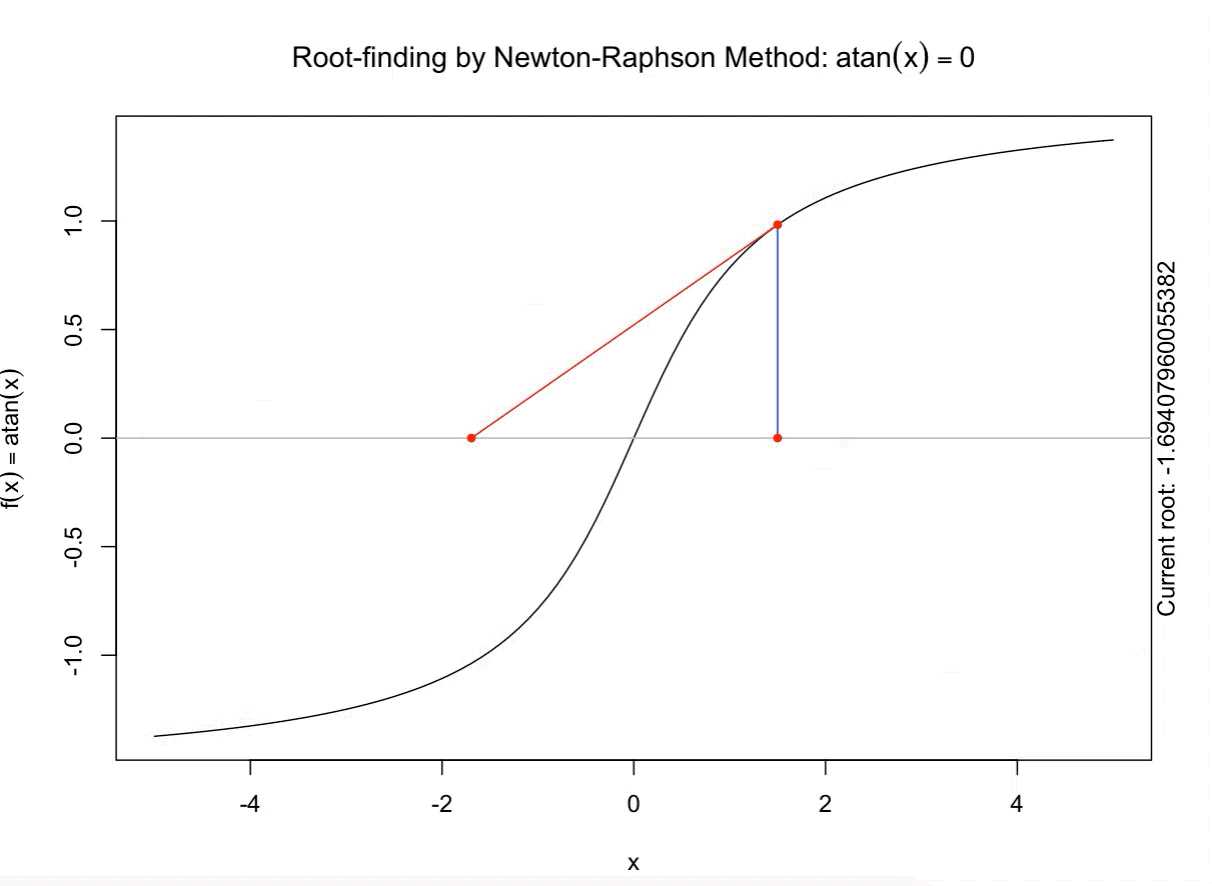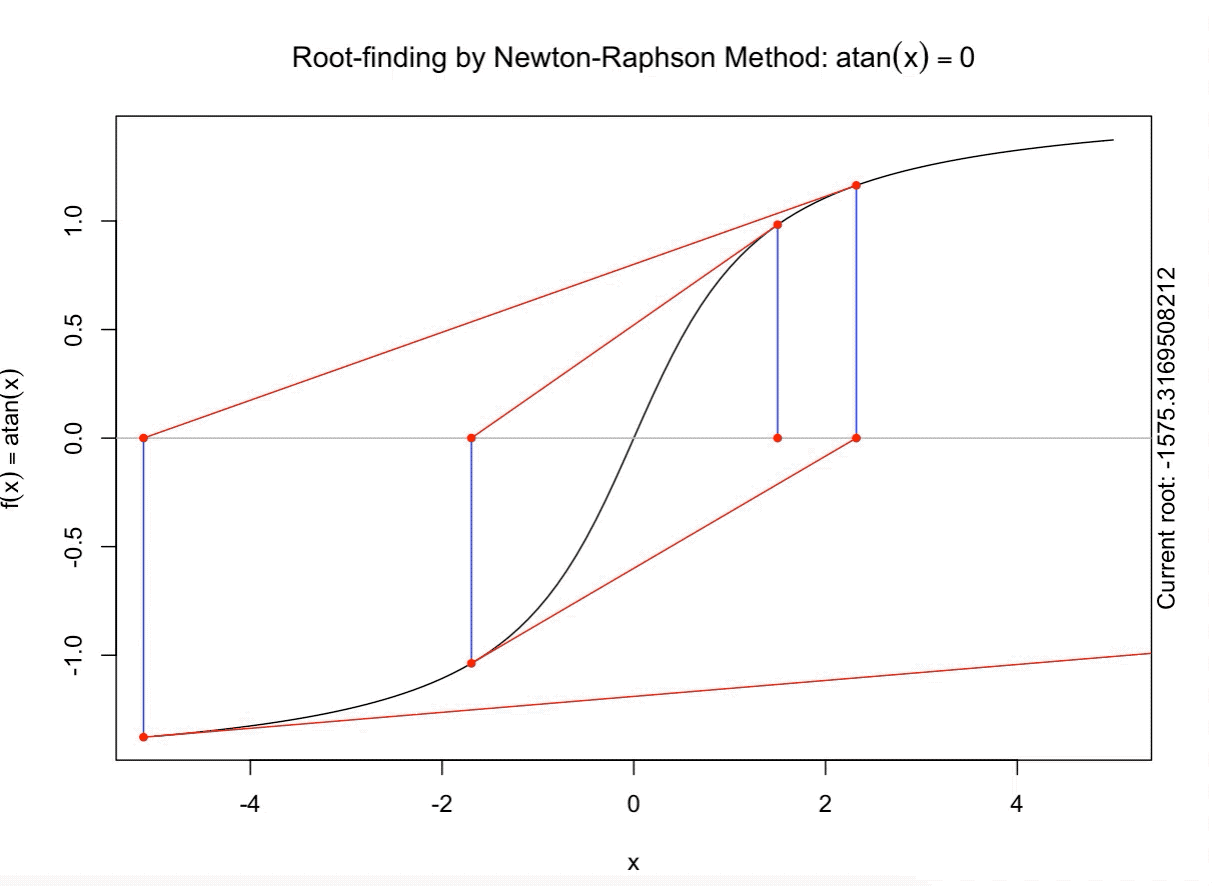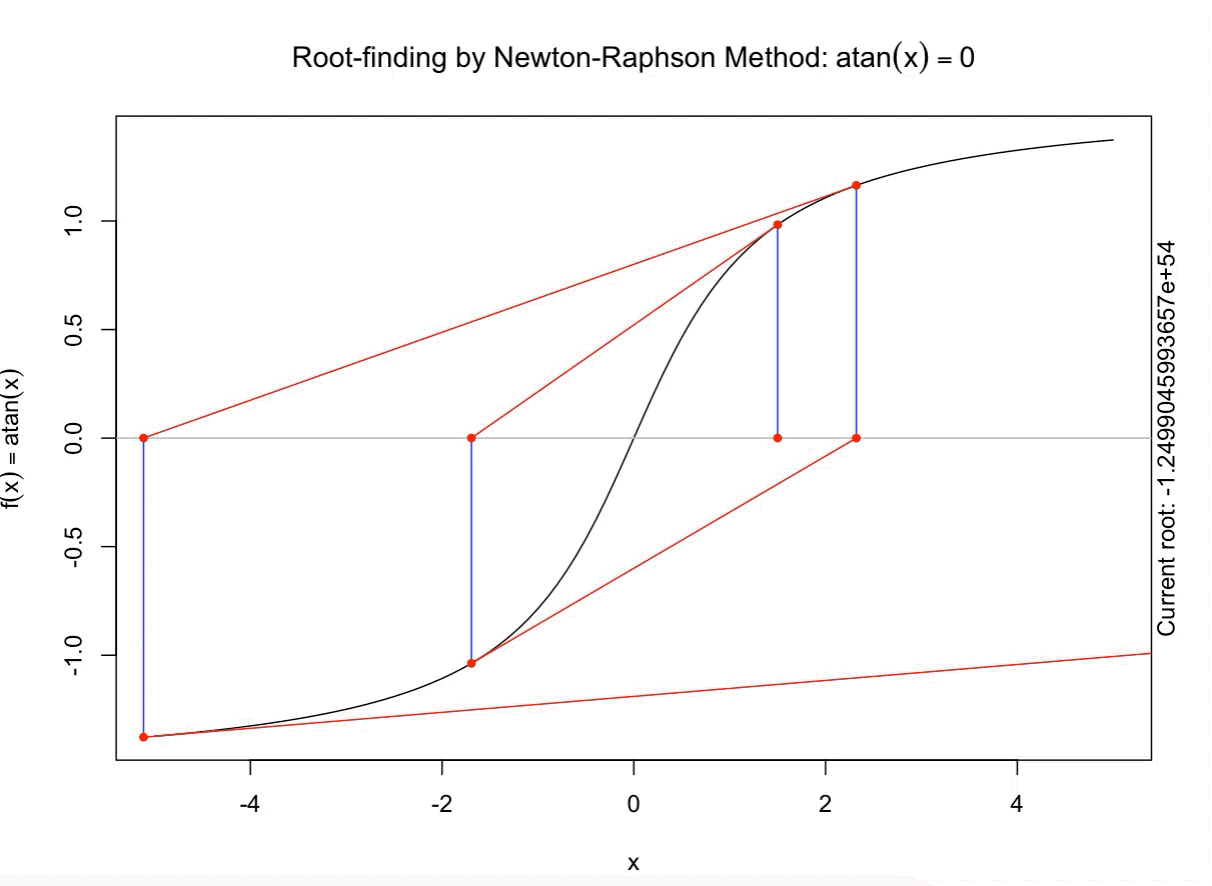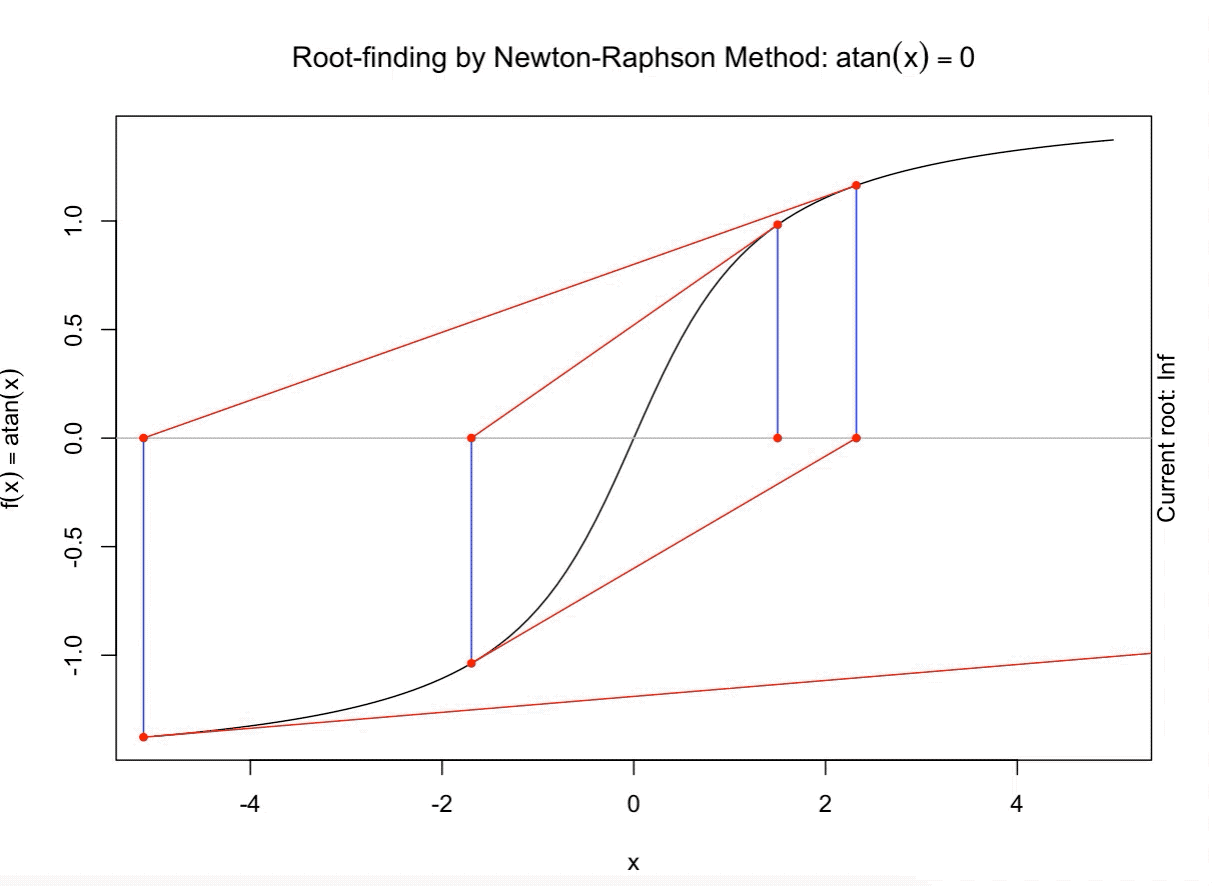] Does not converge. ]] ]]] .column.bg-blue[.content[ ]] --- class: split-60 with-border .column[.content[ # Changing initial values for `asreml` First get a table of initial values: ```r sv <- asreml(Yield ~ Site, data=data1, random= ~Site:Geno, * start.values=TRUE) sv$gammas.table ``` ``` Gamma Value Constraint 1 Site:Geno!Site.var 0.1 P 2 R!variance 1.0 P ``` Modify as you see fit: ```r sv$gammas.table$Value <- c(0.5, 1) fit <- asreml(Yield ~ Site, data=data1, random= ~Site:Geno, * G.param=sv$gammas.table, * R.param=sv$gammas.table) ``` ]] .column.bg-blue[.content[ # Default Values The default initial values are 0.1 for both variance ratios and correlations with some exceptions, e.g. for `us` models it is given by ```r matrix(0.1, nrow=n, ncol=n) + diag(0.05, nrow=n) ``` ### When to change initial values? When you have some better idea of what the parameter values should be, e.g. from past study or estimates from more simpler model going to a more complex model. ]] --- class: split-70 with-border .column[.content[ # Compound Symmetry Model ```r fit <- asreml(Yield ~ Site, data=data1, * random=~ Geno + Site:Geno) lucid::vc(fit) ``` ``` effect component std.error z.ratio constr Geno!Geno.var 1.092 1.252 0.87 P Site:Geno!Site.var 0.0000004 0.0000002 2.5 B R!variance 0.9553 0.3747 2.5 P ``` ```r sv <- asreml(Yield ~ Site, data=data1, random= ~Geno + Site:Geno, start.values=TRUE) sv$gammas.table ``` ``` Gamma Value Constraint 1 Geno!Geno.var 0.1 P 2 Site:Geno!Site.var 0.1 P 3 R!variance 1.0 P ``` ]] .column.bg-blue[.content[ ### Changing constraint to `"U"` ```r sv$gammas.table[,3] <- c("P", "U", "P") ``` The proposed model is the same as: <img src="images/CSmodel.png" width="100%"/> ]] --- class: split-70 with-border .column[.content[ # Allow for unconstrained estimate ```r fit <- asreml(Yield ~ Site, data=data1, random= ~Geno + Site:Geno, G.param=sv$gammas.table, R.param=sv$gammas.table) vc(fit) ``` ``` effect component std.error z.ratio constr Geno!Geno.var 1.099 1.255 0.88 P Site:Geno!Site.var -0.03282 0.3954 -0.083 U R!variance 0.9755 0.4599 2.1 P ``` If you used the code "F" instead, it will fix the parameter to the specified initial value. ]] .column.bg-blue[.content.vmiddle[ ]] --- class: split-two .column.bg-yellow[.content[ # References * Butler et al. (2009) ASReml-R reference manual. [Technical Report.](https://www.vsni.co.uk/downloads/asreml/release3/asreml-R.pdf) ]] .column.bg-main1.white[.content[ # How to get help * For technical help, the best place is to post at the [VSN forum](https://www.vsni.co.uk/forum/). * For sofware installation and license issues, please contact [support@vsni.co.uk](mailto:support@vsni.co.uk). * [Cross Validated](https://stats.stackexchange.com/questions/tagged/asreml) with the tag as `asreml`. The questions are very sparse but I roam around Stack Overflow and Cross Validated and answer occasionally. ]] --- class: split-40 title-slide2 with-border white background-image: url("images/bg2.jpg") background-size: cover .column[.content[ ]] .column.shade_main[.content[ <br><br> # <u>Slides</u> These slides were made using the R package [`xaringan`](https://github.com/yihui/xaringan) with the [`ninja-themes`](https://github.com/emitanaka/ninja-theme) and is available at [`bit.ly/UT-WS-asreml-R`](http://bit.ly/UT-WS-asreml-R). # <u>Your Turn</u> <s>Download `day3-session01-intro-to-asreml.Rmd` here, open in RStudio, push the button "Run Document" on the top tab and work through the exercises.</s> For workshop participants, contact Emi for the tutorials. <br><br> ### <a rel='license' href='http://creativecommons.org/licenses/by-sa/4.0/'><img alt='Creative Commons License' style='border-width:0; width:30pt' src='images/cc.svg' /><img alt='Creative Commons License' style='border-width:0; width:20pt' src='images/by.svg' /><img alt='Creative Commons License' style='border-width:0; width:20pt' src='images/sa.svg' /></a><span style='font-size:10pt'> This work by <span xmlns:cc='http://creativecommons.org/ns#' property='cc:attributionName'>Emi Tanaka</span> is licensed under a <a rel='license' href='http://creativecommons.org/licenses/by-sa/4.0/'>Creative Commons Attribution-ShareAlike 4.0 International License</a>.</span> ]] <img src="images/USydLogo-white.svg" style="position:absolute; top:80%; left:80%;width:200px">